|
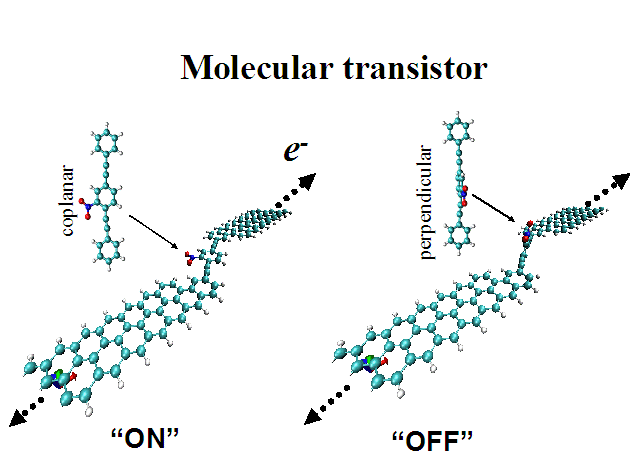
We have performed ab initio calculations of electronic and transport properties of nano-electronic systems. The surface Green’s function of semi-infinite ribbons made from graphene sheets is constructed from ab initio one-dimensional bulk calculations, and the energy dependent transmission coefficient through a molecular tunneling junction is computed from Landauer’s formalism within a framework that combines Caroli’s approach and density functional theory. By using the same level of theory and basis set to model both the molecule and the ribbons, and by an exact, analytical determination of surface Green’s functions, we aim for a quantitative description of the configuration dependent electron transport through ribbon-molecule-ribbon switching devices.
|
|
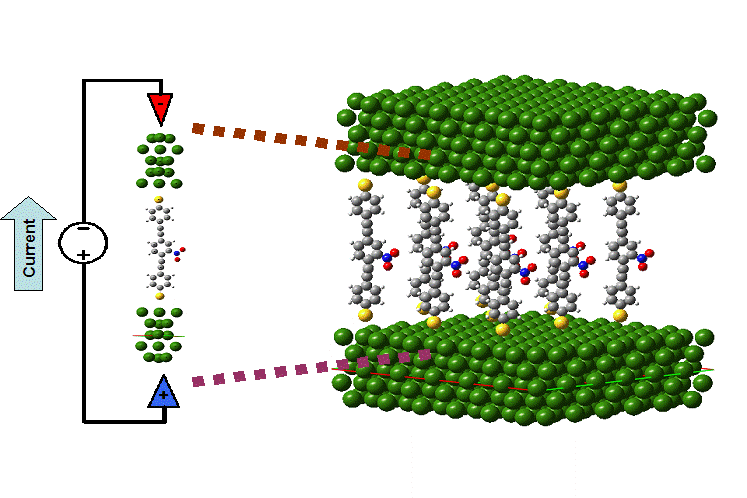
Second, we are currently implementing a generalization of the previous work. This formalism will allow us to calculate electron transport through 2-dimensional nano-electronic systems, such as the transport through self-assembled monolayers.
|
|
Third, we are exploring the use of Quasiatomic Minimal Basis Orbitals (QUAMBOs) [Lu et al, Phys. Rev. B 70, 041101 (2004)] as the default basis set to our transport formalism. A QUAMBO basis set will allow scientists using plane-wave-based codes (i.e. VASP, PWscf, DACAPO, etc) to use our formalism and computer programs. Also, because of the good scalability of commercially available plane-wave codes, the use of QUAMBOS will make possible the study of larger nano-electronic systems than currently possible with atomic-orbital-based commercial packages. |